表1临床表现及体征
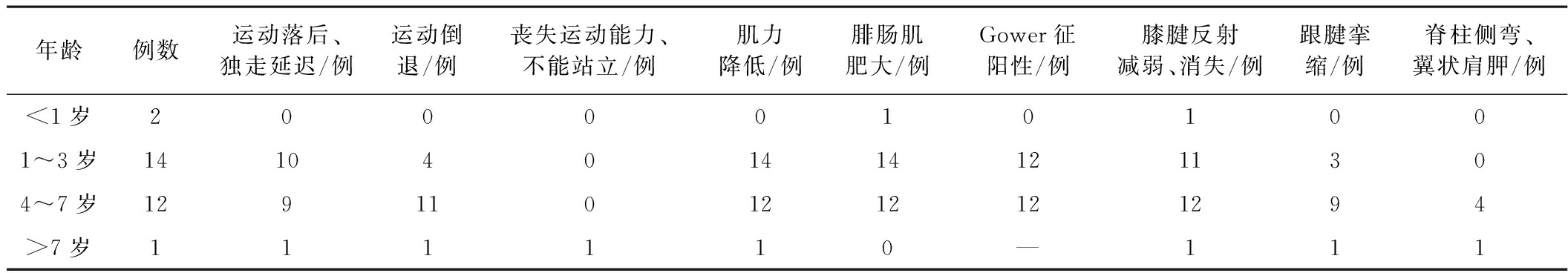
年龄例数运动落后、独走延迟/例运动倒退/例丧失运动能力、不能站立/例肌力降低/例腓肠肌肥大/例Gower征阳性/例膝腱反射减弱、消失/例跟腱挛缩/例脊柱侧弯、翼状肩胛/例<1岁20000101001~3岁14104014141211304~7岁1291101212121294>7岁111110—111
王燕娟,胡文广,刘 平,邓 佳,赵力立
(成都市妇女儿童中心医院,四川 成都 610091)
【摘要】目的:分析29例Duchenne型肌营养不良(Duchenne muscular dystrophy,DMD)的临床表现和基因分型,为早期诊断提供可靠方法。方法:纳入2011年1月至2016年12月于我院基因明确诊断DMD的患儿共29例,回顾性分析其临床表现,包括肌力、运动功能、肌酸激酶和心电图、肌电图、心功能,同时通过多重连接依赖式探针扩增(multiplex ligation-dependent probe amplification,MLPA)和第二代基因测序技术的方法进行基因分析。结果:DMD以男性发病为主,多隐匿起病,进行性肌无力,近端重于远端,伴腓肠肌肥大,10~12岁左右失去运动能力,并逐渐出现心肺功能下降。29例患儿肌酸激酶均明显升高,肌电图示肌源性损害。29例患儿均进行MLPA及二代测序检测,其中19例为外显子缺失(65.52%),5例为外显子重复(17.24%),5例为点突变(17.24%),新发突变占37.93%。结论:认识DMD的临床特点,及时进行基因检查,有助于提高该病的临床诊断水平,早期合理治疗,可避免误诊误治,并有利于遗传咨询。
【关键词】Duchenne型肌营养不良;临床表现;基因分析;MLPA;第二代基因测序
Duchenne型肌营养不良(Duchenne muscular dystrophy,DMD)也称杜氏肌营养不良或假性肥大型肌营养不良,是最常见的X连锁隐性遗传的肌肉病。发病率约为1/3500活产男婴[1]。该病多隐匿起病,在出生约18个月后开始行走,呈鸭步,容易摔倒,不能正常跑,上楼梯困难,不能双足或单足跳跃。3~5岁出现症状,运动功能呈进行性倒退,四肢近端肌萎缩,鸭步加重,下蹲不能站起,上梯更加困难。体征可见双侧小腿腓肠肌肥大,膝腱反射减弱或消失,Gower征阳性,逐渐出现跟腱挛缩、翼状肩胛、脊柱侧弯。6~8岁逐渐出现心功能异常,10~12岁左右逐渐丧失独立行走能力,需要依赖轮椅,膝、髋、肘关节出现挛缩,脊柱侧弯更加明显,腓肠肌逐渐萎缩。多于20岁左右因心功能不全和(或)呼吸衰竭死亡,部分患儿还伴有智力低下。该病是定位于Xp21.1区的抗肌萎缩蛋白基因(dystrophin,也称DMD基因)缺陷引起肌细胞膜骨架蛋白缺失,导致肌细胞膜完整性破坏[2]。确诊DMD主要依靠基因检查,另外肌电图、骨骼肌MRI及肌肉活检可以辅助明确诊断[3]。由于本病目前缺乏有效的根治方法,因此早期诊断有助于早期治疗,更好地进行遗传咨询,控制和降低DMD发病率。
1.1 研究对象 选择2011年1月至2016年12月于我院小儿神经科就诊,临床及基因确诊DMD的患儿。共29例患儿,就诊年龄5个月至11岁,均为男性,病程2个月至5年。其中3例(11.1%)有母系家族史, 1例是患儿的同胞哥哥,另外2例是患儿的舅舅。
1.2 方法 制定统一的调查表搜集所有患儿的临床资料。在征得监护人知情同意后,所有患儿均送康旭基因公司进行了DMD基因检查,包括通过MLPA技术检测DMD基因79个外显子缺失/重复的情况及通过第二代基因测序技术检测点突变的情况。
2.1 临床表现 29例患儿均为男性。其中2例<1岁的婴儿仅表现为体检时发现肌酸激酶升高,肢体活动基本正常。其他患儿起病隐匿,绝大部分患儿1岁后出现运动较同龄儿落后,独走时间延迟,易跌倒(21/27,占77.8%);4~5岁出现行走缓慢,呈典型鸭步,重者不能跑跳、上楼梯困难、蹲起费劲;超过10岁的患儿已丧失行走能力。体格检查:肌肉萎缩,尤以双下肢近端肌肉为著,腓肠肌假性肥大,双下肢肌力下降,肌张力降低,膝腱反射消失,跟腱挛缩,Gower征阳性,可见翼状肩胛。经智力运动评估,其中6例(20.7%)患儿有不同程度的精神发育迟滞。29例患儿中的5例(17.4%)曾在院外误诊为病毒性肝炎,经过1个月至1年的保肝治疗。见表1。
表1临床表现及体征
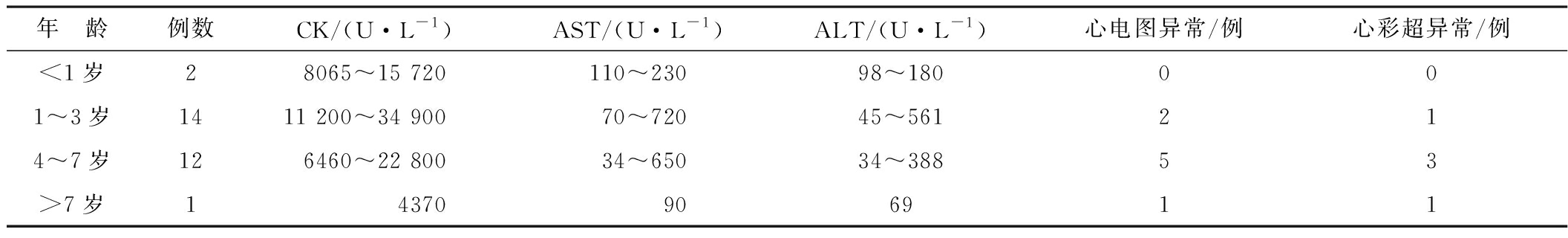
2.2 辅助检查 (1)实验室检查:肌酸激酶(CK)均明显升高,为4370~34 900 U/L,为正常参考值20~100倍,同时伴肝酶升高,以谷草转氨酶(AST)、谷丙转氨酶(ALT)升高为主。(2)心电图:8例异常,其中3例ST段、T波改变,1例多导联高尖P波,2例完全/不完全房室传导阻滞,2例左室高电压。(3)心彩超:5例异常,其中2例示少量三尖瓣反流;1例示二尖瓣关闭不全;2例示左心室扩大,其中1例伴心功能减低;其他均为正常。(4)肌电图:其中15例完成肌电图,均提示肌源性损害,1例伴有神经传导速度减慢。(5)骨骼肌MRI:1例行骨骼肌MRI,示臀肌及大腿肌肉不同程度萎缩,脂肪浸润。(6)肌肉活检:1例行肌肉活检,所检肌肉为腓肠肌,表现为肌纤维大小不一,部分坏死和再生,结缔组织增生。见表2。
表2辅助检查结果
2.3 基因分析结果 所有患儿行DMD基因MLPA及二代基因测序检测,其中19例检测出外显子缺失,占65.52%;5例检测出外显子重复,占17.24%;另外5例检测出点突变,占17.24%。以外显子45~53缺失最为常见;新发突变占37.93%。
Duchenne型肌营养不良的主要临床特征是进行性肢体肌萎缩肌无力,近端为主,伴小腿腓肠肌假性肥大。一般3~5岁出现症状,呈进行性加重,10~12岁丧失行走能力,多于20岁左右死于呼吸衰竭和(或)心力衰竭。研究中的患儿临床进程基本符合DMD的临床特点,值得注意的是相当部分患儿有运动发育迟滞,但常被家长忽略,而经仔细追问,高达77.8%的患儿均有独走的延迟。提示临床医生,在发现不明原因独走延迟的男性患儿时应考虑到DMD的可能性。
DMD患者出生后即可出现肌酸激酶升高,常为正常的10倍以上,到3岁左右达高峰,是正常的50~100倍,然后随着越来越多的肌肉被脂肪代替和纤维化,肌酸激酶水平逐渐下降[4]。因此,婴幼儿早期发现不明原因肌酸激酶增高的男性患儿时,应警惕DMD的可能。此外,DMD患儿在肌酸激酶明显增高时常会伴有肝酶升高,当患儿尚没有明显肌病症状体征和(或)临床医生忽略了肌病相关症状体征筛查时,可能被误诊为病毒性肝炎[5]。本研究中患儿被误诊达5例(17.4%),经长达1个月至1年的保肝治疗,延误了正确诊断的时间。DMD患者常合并心脏损害,心电图是最简单可靠的方法,心电图异常出现率高且较为突出和特异性的改变为:胸部导联 (V16)和肢体导联 (Ⅰ、Ⅲ、 AVL、AVF)窄而深 Q波、高尖的P波和R波,各种类型传导阻滞、期前收缩、心动过速等也很常见[6]。超声心动图可提示左心室扩大,心功能下降。肌电图均提示肌源性受损,也有少数患儿合并周围神经传导速度减慢,远段潜伏期延长或波幅降低,这是因为肌纤维严重损害时可继发神经退行性变[7]。但是由于肌电图只能帮助定位诊断,而不能定性诊断,并且为有创性的检查,在儿科的应用有所受限。肌肉MRI作为无创检查,能显示受累肌肉出现不同程度的水肿、 脂肪浸润和间质增生[8],在肌病诊断中的应用日益增多。肌肉活检可显示肌细胞大小不等,出现坏死、再生,以及被脂肪和结缔组织替代,免疫组化染色提示Dystrophin蛋白完全或严重缺失[9],这也是DMD特异性的诊断指标[10,2]。
DMD是位于X染色体上编码生成抗肌萎缩蛋白的基因发生缺陷所致。抗肌萎缩蛋白基因是迄今发现的最大的人类基因,具有79个外显子,大约230万个碱基对,其蛋白质产物也非常大,分子量为427 000。DMD基因具有极高的突变频率,主要分为3种突变类型:外显子缺失、外显子重复和点突变。其中外显子缺失占55%~65%,重复占5%~15%,点突变占30%左右,主要的缺失热区位于外显子45~54之间,占总缺失的54%~60%[11],基因突变类型中约30%是新发突变[12],即非遗传所致。本研究的基因分型发现19例外显子缺失,占65.52%;5例外显子重复,占17.24%;5例点突变,占17.24%。以外显子45~53缺失最为常见,新发突变占37.93%,与既往研究基本符合。MLPA能检测外显子缺失/重复突变,但不能检测出点突变;而第二代基因测序技术可检测出点突变,但不能检测出外显子大片段缺失和重复突变,所以临床进行DMD基因检测时,需要结合MLPA及基因测序,才能更全面检测出基因突变的类型。另外,研究表明,这些新生突变虽然母亲未发现基因突变,但实际可能是生殖细胞或体细胞的嵌合现象,其后代中有5%~10%的概率也会携带相同的突变,所以在妊娠后应接受产前诊断,其女性亲属也应接受携带者的检测[13]。随着基因检查技术的不断发展,在实际诊断流程中,当临床考虑DMD时应首选基因检查,能够更高效、准确、无创地进行诊断。而基因未检出异常或需要鉴别是DMD还是BMD(Becker型肌营养不良)时,才进行有创性的肌活检。
本研究中,患者的临床进展符合DMD的自然病程,个别伴有家族史,体格检查可见典型的腓肠肌肥大,膝腱反射减弱或消失,Gower征阳性,逐渐出现跟腱挛缩,查肌酸激酶均有明显升高,伴有肌源性损害肌电图的表现或肌肉MRI显示肌肉不同程度萎缩、脂肪浸润,随着年龄的增加,部分患儿因心脏受累,心电图及心彩超出现异常改变,临床即可考虑诊断DMD。所有患儿的基因检查结果最终证实了临床诊断。
目前DMD尚无特效的治疗,药物治疗以糖皮质激素为主[14-15],其他包括营养、呼吸、心脏、矫形、康复、心理等综合治疗[16],可改善DMD患儿肌力和肌肉功能,延长行走能力3~5年,增强心肺功能,减少脊柱畸形,延长生命期,提高生存质量。另外,近年来随着外显子跳跃疗法的研究成功[17-18],将有望成为治疗本病的新方法,但需长时间对其安全性和有效性进行观察和验证后,才能进入真正的临床治疗阶段。
DMD是一种慢性进展性致死性疾病,西南地区经济欠发达,家长对患儿运动发育落后的认识不足,常常导致诊断的延误;部分临床医生对本病认识不足,更易导致误诊误治。应提高对本病的认识,包括临床的症状、体征、病情的自然进程及辅助检查的特点,以期早诊断、早治疗,延缓患儿病情进展,提高其生活质量并延长生存期。同时以便于更好地进行遗传咨询和计划生育,对产前诊断及携带者的检测有非常重要的意义,可减少群体遗传负荷,也可提高优生优育的质量,控制和降低DMD发病率。
参考文献:
[1]EMERY A E. Population frequencies of inherited neuromuscular diseases-a world survey[J]. Neuromuscul Disord, 1991, 1(1): 19-29.
[2]胡静.骨骼肌疾病临床病理诊断 [M].北京:人民卫生出版社,2011:6-72.
[3] 中华医学会神经病学分会.中国假肥大型肌营养不良症诊治指南[J].中华神经杂志,2016,49(1):17-20.
[4]王丽波,麻宏伟,王琳,等.Duchenne肌营养不良患儿的智力特点及与基因突变关系初步探讨[J].中国当代儿科杂志,2011,13(10):804-807.
[5]刘平,吴惧,胡文广,等.儿童进行性肌营养不良误诊为病毒性肝炎5例临床分析[J].临床误诊误治,2012,25(7):40-42.
[6]李伟,卢锡林,梁银杏,等.假肥大型肌营养不良心脏功能改变(附406例分析)[J].中国临床神经科学,2010,18(1):29-33.
[7]刘敏,石靖.进行性肌营养不良患儿的临床及肌电图分析[J].临床神经电生理学杂志,2008,17(6):340-342.
[8]王霞,赵初青,王文涛,等.MRI对Duchenne肌营养不良的诊断价值[J].实用放射学杂志,2016,32(9):1414-1421.
[9]喻绪恩,王训,石永光,等.Duchenne型肌营养不良的临床和病理及抗肌萎缩蛋白表达[J].中国临床神经科学,2012,20(2):153-158.
[10]MENDELL J R, CAMPBELL K, RODINO-KLAPAC L A, et al. Brief report: dystrophin immunity in Duchenne’s muscular dystrophy[J]. N Engl J Med, 2010, 363(15): 1429-1437.
[11]MILLER R G,HOFFMAN E P.Molecular diagnosis and modern concern management of Duchenne muscular dystrophy[J].Neurol Clin,1994,12:699-725.
[12]0U Z, LI S, LI Q, et al.Duchenne muscular dystrophy in a female patient with a karyotype of 46,X,i(X)(q10)[J]. Tohoku J Exp Med, 2010, 222(2): 149-153.
[13]戚庆炜,孙念怙,郝娜.应用荧光原位杂交技术诊断基因缺失型进行性假肥大性肌营养不良携带者[J].中华医学遗传学杂志,2003,8(4):82-84.
[14]高洪杰,付溪,罗小平.进行性肌营养不良的糖皮质激素治疗现状及进展[J].中华儿科杂志,2015,53(6):476-479.
[15]GLOSS D, MOXLEY I R, ASHWAL S, et al. Practice guideline update summary: corticosteroid treatment of Duchenne muscular dystrophy report of the Guideline Development Subcommittee of the American Academy of Neurology[J]. Neurology, 2016, 86(5): 465-472.
[16]胡君,蒋莉.Duchenne型肌营养不良的诊治与管理,2:多学科协作模式[J].儿科药学杂志,2012,18(8):41-48.
[17]李亚勤,张成.外显子跳跃剪接法治疗Duchenne型肌营养不良的机制研究进展[J].中华神经科杂志,2011,44(6):419-422.
[18]BHAGAVATI S.Exon-skipping therapy for Duchenne muscular dystrophy[J]. Lancet, 2012, 379(9811): 10-11.
WANG Yanjuan,HU Wenguang,LIU Ping, DENG Jia,ZHAO Lili
(Chengdu Women and Children’s Central Hospital,Chengdu, Sichuan 610091,China)
【Abstract】Objective: To analyze the clinical manifestations and genotyping of 29 cases of Duchenne muscular dystrophy (DMD); and to provide a reliable method for early diagnosis.Methods: A retrospective analyze was made in the clinical manifestations of 29 children diagnosed as DMD in our hospital from January 2011 to December 2016, including muscle strength, motor function, creatine kinase and ECG, electromyography and cardiac function. Gene analysis was conducted through multiplex ligation-dependent probe amplification (MLPA) and second-generation sequencing technology.Results: DMD was a male-dominated disease. It showed clinical manifestations of most hidden onset, progressive myasthenia, heavy symptoms at the proximal end compared with the distal end, the gastrocnemius muscle hypertrophy, the loss of motor ability around 10-12 years old, and the gradual decline of cardiopulmonary function. Creatine kinase was elevated, and electromyography showed myogenic damage in 29 cases. They all received MLPA and second-generation sequencing detection, including 19 cases of missing exons (65.52%), 5 cases of repeated exons (17.24%), 5 cases of point mutations (17.24%) and 11 cases of new mutations (37.93%).Conclusion: Understanding the clinical features of DMD and carrying out gene examination timely is useful for the improvement of clinical diagnosis, early reasonable treatment, the avoidance of misdiagnosis and mistreatment, and genetic counseling of this disease.
【Keywords】Duchenne muscular dystrophy; clinical manifestations; genetic analysis; MLPA; second generation of gene sequencing
【中图分类号】R746.2
【文献标识码】A
DOI:10.11851/j.issn.1673-1557.2018.04.018
优先数字出版地址:http://kns.cnki.net/kcms/detail/51.1688.R.20180723.1554.018.html
通信作者:刘平,35402865@qq.com
(收稿日期:2017-06-08)